
Displaying SNPs and indels
In this tutorial we’ll cover using Circleator to display SNPs and indels between closely-related strains. Before proceeding with the tutorial, please make sure that you have Circleator installed as described in the Circleator Installation Guide.
Outline
-
Example 1: Reading single-sample variants from VCF files
Reading single-sample variants from VCF files
Download the input and configuration files
In this first example we’ll look at displaying variants (SNPs, insertions, and deletions) from VCF files. We’re going to use the same reference genome, that of Enterobacteria phage lambda, as in Examples 1 and 2 from the Coverage Plots tutorial. In that tutorial we used Bowtie2 to align the synthetic read data provided with Bowtie against the phage lambda genome, as described in the Bowtie2 documentation. The Bowtie2 documentation also describes how to call variants and generate a VCF file with the samtools package and one of the BAM alignment files. We’ll use that VCF file for our first Circleator example. There’s just one small difference between the samtools command in the Bowtie documentation and the one that we’re going to use, because we want to generate a “.vcf” file rather than a “.bcf” file (i.e., a plain VCF file rather than a binary-encoded VCF file.) So instead of the following command, from the Bowtie2 documentation:
samtools mpileup -uf lambda_virus.fa eg2.sorted.bam | bcftools view -bvcg - > eg2.raw.bcf
We will instead use this command (with no -b flag and “.vcf” instead of “.bcf”):
samtools mpileup -uf lambda_virus.fa eg2.sorted.bam | bcftools view -vcg - > eg2.raw.vcf
Here is the VCF file that this command should produce, assuming that it’s
run on the eg2.sorted.bam file from the Coverage Plots tutorial:
Here is the GenBank flat file for the phage lambda genome:
Here is the contig list file that reads the GenBank flat file but which tells Circleator to use
“gi|9626243|ref|NC_001416.1|” as the id for the sequence that it contains, instead of “NC_001416”.
This file is passed to Circleator’s --contig_list option, rather than passing the GenBank flat
file directly to the --data option. Without this indirection Circleator won’t be able to match
the variants in the VCF file (which are looking for a sequence with id “gi|9626243|ref|NC_001416.1|”,
not “NC_001416”) with the reference sequence. This id-matching issue is also described in the
Coverage Plots tutorial.
And here is the Circleator configuration file for our first figure:
Run Circleator
Once you’ve downloaded or generated the necessary files, you’re ready to run Circleator, like so:
$ circleator --contig_list=contig-list-ex1-gb.txt --config=variants-ex1.txt > variants-ex1.svg
Convert the figure from SVG to PNG
If everything looks good so far then use rasterize-svg to convert the SVG to a PNG file:
$ rasterize-svg variants-ex1.svg png 3000 3000
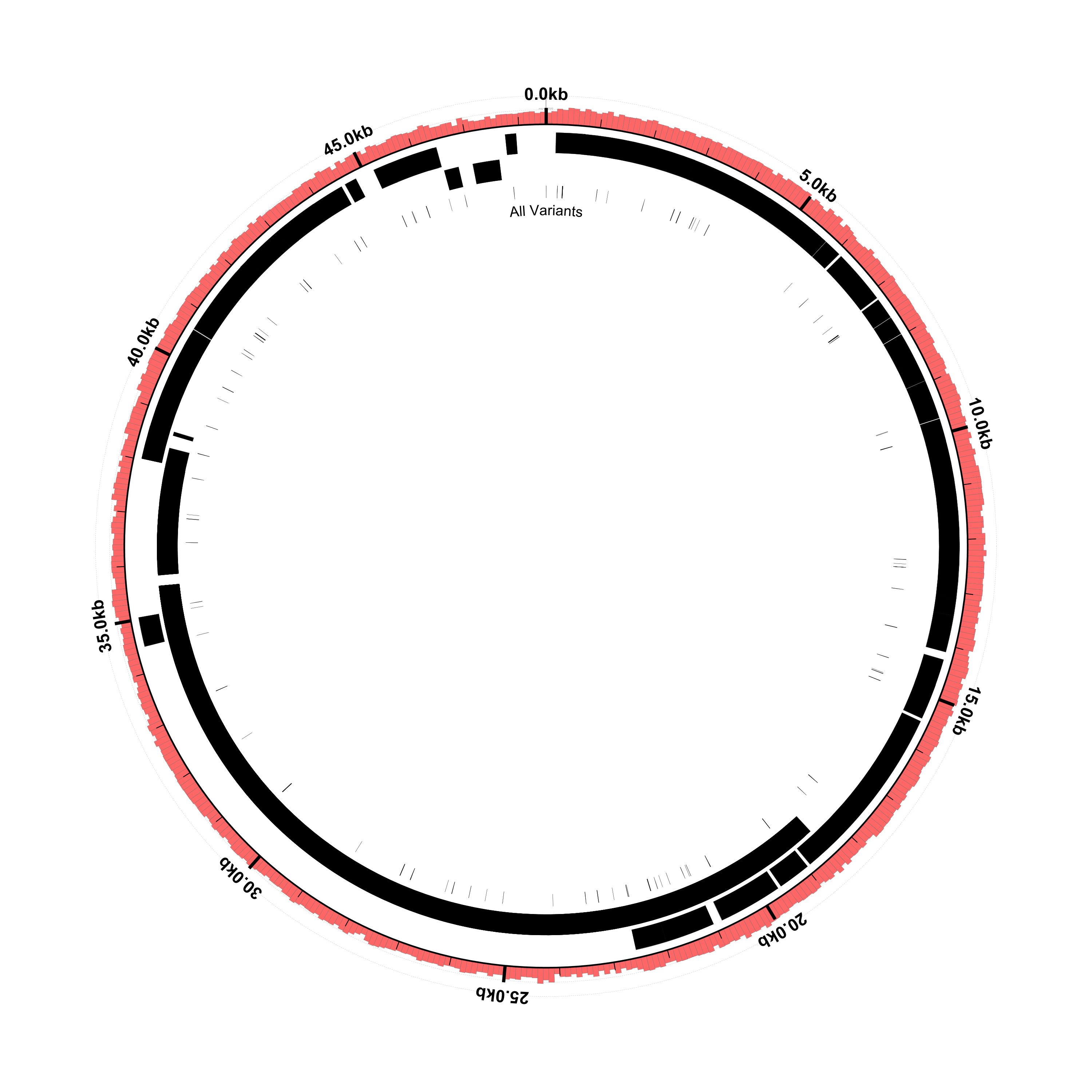
Here’s what the result should look like:
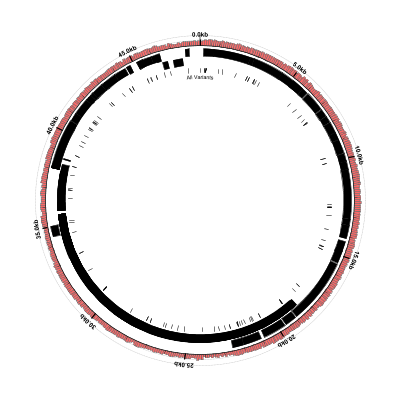
Blow things up
This type of figure is useful for visualizing where the SNPs and indels tend to cluster, but does not provide
as much detail as the VCF file. One thing we can do is “blow up” or expand the variant loci (i.e., manipulate
the scale of the figure around the circumference of the circle so that the regions of interest are expanded or
stretched and all the other regions are compressed accordingly.) We can do this with the scaled-segment-list
glyph in Circleator and, once the scale has been changed, we will have more space to overlay useful information
on the variant loci. We’ll use the same input contig list file, GenBank flat file, and VCF file, but the
following modified Circleator configuration file:
Save the above file and rerun Circleator, replacing the original configuration file with the new one (and changing the name of the output SVG and PNG files):
$ circleator --contig_list=contig-list-ex1-gb.txt --config=variants-ex1b.txt > variants-ex1b.svg
$ rasterize-svg variants-ex1b.svg png 3000 3000
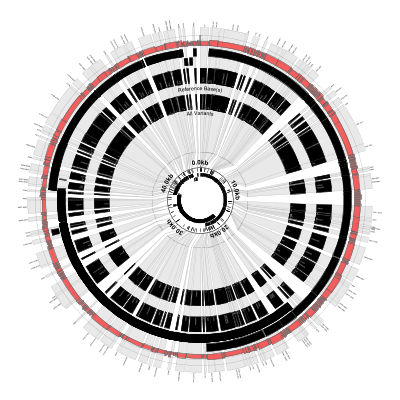
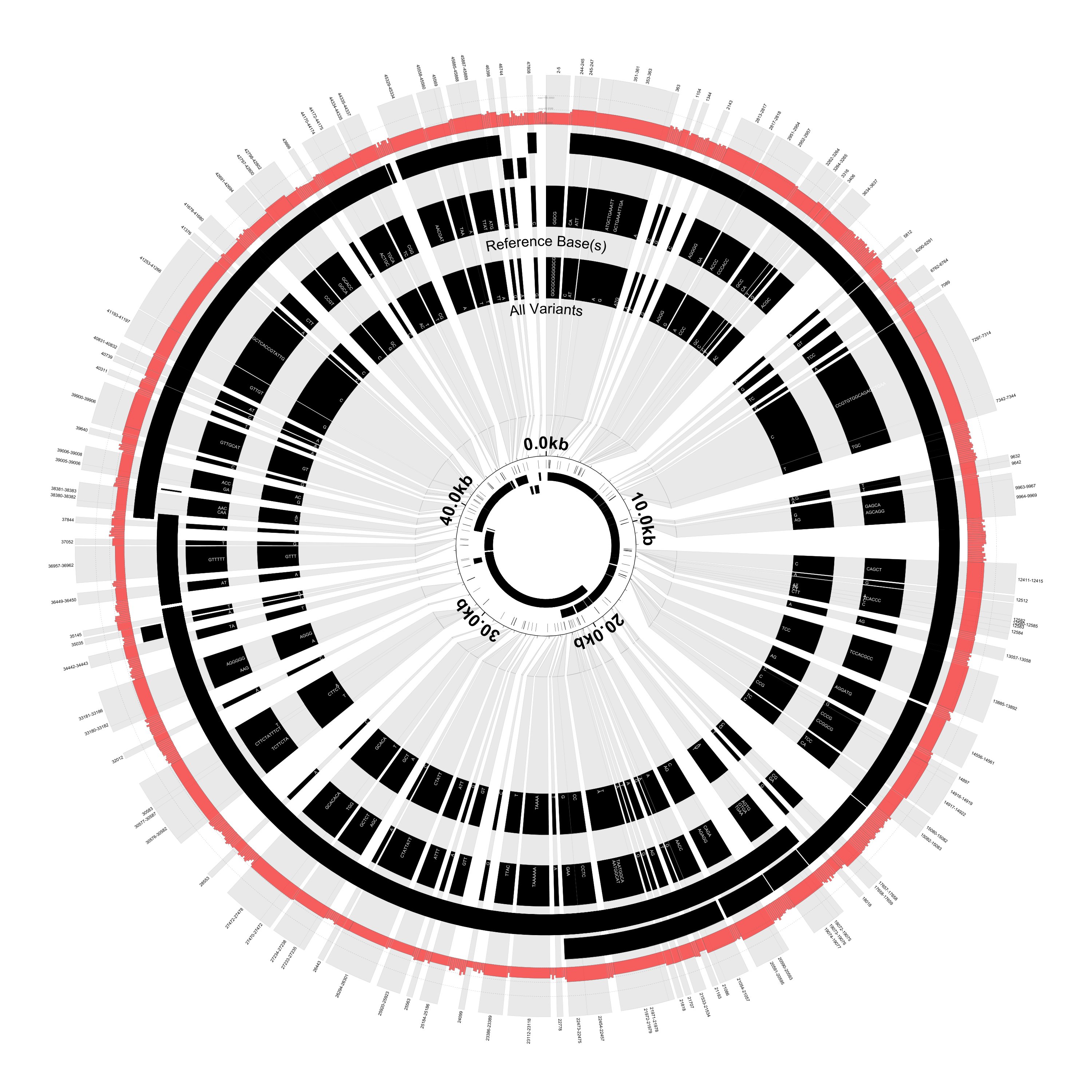
In this version of the figure, notice that:
- Each variant locus is shaded grey and expanded by about 100X
- The coordinate labels have been relocated to the inside of the figure
- The reference and variant base(s) are now overlaid on the figure
- Each variant locus is labeled with its base position (the outermost part of the figure)
- The small inner circle shows the genes and SNPs without any scaling
- Some of the called variants overlap (e.g., position 12580-12585)

{kind=link}
{kind=link}
{kind=link}
{kind=link}