
Circleator - Configuration File Reference
Introduction
The Circleator configuration file, specified by the --config command line
option, determines almost every aspect of how Circleator will draw the figure.
This document describes the configuration file format in detail, and covers
the following topics:
- File Format
- Predefined Tracks
- User Defined Tracks
- Track Size and Position
- Feature Selection
- Colors
- Labels
- Opacity
- Loops
- Z-Index
File Format
The Circleator configuration file is a tab-delimited plain text file designed to be edited manually. Blank lines or lines beginning with the “#” character (similar to Perl-style comments) are ignored. Every other line must contain at least one of the following tab-delimited fields (almost all of which are optional):
- type - either the name of a predefined track type OR the keyword
new - name - a name by which the track may be referenced from elsewhere in the configuration file
- glyph - the Circleator “glyph” used to render this track
- heightf - the height of the track as a fraction of the circle’s radius (0-1)
- innerf - position of the innermost part of the track as a fraction of the circle’s radius (0-1)
- outerf - position of the outermost part of the track as a fraction of the circle’s radius (0-1)
- data - path to a data file, if one is required by the chosen track type and/or glyph
- feat_type - display only features of the specified type (e.g., “gene”, “tRNA”)
- feat_strand - display only features on the specified strand (e.g., “-“, “+”, “-1”, “1”)
- color1 - interpretation depends on the track type: usually the SVG fill color
- color2 - interpretation depends on the track type: usually the SVG stroke (outline) color
- opacity - opacity of the track between 0 and 1, where 0 = invisible/completely transparent and 1=completely opaque
- zindex - integer z-index of the track: tracks with higher z-indexes are drawn on top of those with lower z-indexes
- options - a comma-delimited list of track options in the format “key=value”
All of these fields except for the very first one (type) are optional and may be omitted completely. Hence the simplest possible line/track in a Circleator configuration file is the name of a predefined track type and nothing else, like this:
coords
You can choose to include as many or as few of the field values as you like, provided that the type is always specified. But note that once you start skipping fields, you have to skip all the rest too. So, for example, you could choose to include only fields 1-3 (type, name, glyph)
new r1 rectangle
Or you could specify only fields 1-6 (type, name, glyph, heightf, innerf, outerf):
new r1 rectangle null 0.7 0.75
But you cannot specify only fields 1-3 and 10 (type, name, glyph, color1) without including all the blank fields between numbers 3 and 10:
new r1 rectangle #ff0000
The one very important exception to this rule is that you are allowed to include the final options field, even if you’ve skipped some of the fields that precede it. For example, you can specify fields 1-3 plus the final options field (#14):
new r1 rectangle stroke-width=3
Another useful feature is that any of the positional fields listed
above, with the exception of the first and last, can be included (by
name) in the final options field. So while you can’t specify
values for fields 1-3 and 10 like this (i.e., with only a single tab
between rectangle and the color specifier #ff0000):
new r1 rectangle #ff0000
You can do it like this:
new r1 rectangle color1=#ff0000
Or, equivalently, like this (note the lack of spaces between the different options in the comma-separated options list: the configuration file parser does not allow spaces to be used here):
new name=r1,glyph=rectangle,color1=#ff0000
Note that in one of the preceding examples we used the word null to
indicate a missing field value. Here are some other ways to indicate
missing/absent field values:
- Leaving the field–and all those that follow it–out of the line and skipping directly to the options field.
- Using one of the following equivalent ways to indicate a null/undefined value:
null,undef,n/a,na,n,.(period),-(single dash)
Predefined Tracks
(relevant options: type)
If the value in the first column of a line in the configuration file
is anything other than new then it is the name of a predefined track
type. The entire set of Circleator-supported predefined track types
is listed on the predefined tracks page, along with
information on the most commonly-used configuration options for each
track type.
User Defined Tracks
(relevant options: type, name, glyph)
If the value in the first column of a line in the configuration file
is new then the track is a user-defined track and must include
columns 2 (name) and 3 (glyph), either as positional fields or
named values in the options field. Here are the
currently-supported Circleator glyphs:
- ruler
- rectangle
- label
- graph
- scaled-segment-list
- load
- load-bsr
- bsr
- load-trf-variation
- load-gene-expression-table
- load-gene-cluster-table
- compute-deserts
- compute-graph-regions
- cufflinks-transcript
- synteny-arrow
- loop-start
- loop-end
The prdefined track types are all defined in terms of these glyphs, so a predefined track type is really nothing more than an alias for a user-defined track (plus possibly some default values for some of the track options). The coords track, for example, is defined like this in the system-wide predefined track configuration file (conf/predefined-tracks.cfg):
new coords ruler 0.02 tick-interval=100000,label-interval=500000,label-type=curved
This means that placing either of the following two lines into a configuration file will have exactly the same effect:
# use predefined track type alias for the 'ruler' glyph:
coords
# create user-defined 'ruler' track with the exact same options:
new c1 ruler 0.02 tick-interval=100000,label-interval=500000,label-type=curved
Something to note here is that the system-wide predefined track
configuration file is not the only place where new track types can be
defined: every time you create a user-defined track and give it a
unique name (e.g., c1 in the above example) that name can then be
reused in the same configuration file (strictly after the line on
which it first appears) as though it were a predefined track
type. This can be useful in cases where you wish to define a number of
very similar tracks but without having to retype (or cut and paste)
all the track options. For example, in this configuration file
excerpt one of the user-defined tracks is given the name
lightblue_ring and then that name is used later in the same file to
produce another copy of the same track:
genes
small-cgap
new lightblue_ring rectangle feat-type=contig,color1=blue,opacity=0.2,heightf=0.05
small-cgap
# "lightblue_ring" is now an alias for the track defined above
lightblue_ring
Currently the best way to begin learning about the underlying glyphs is to examine the file that defines the predefined track types using the glyphs. This file, called predefined-tracks.cfg, is itself a Circleator configuration file, albeit with a significant amount of documentation and special processing directives embedded in its comments.
Track Size and Position
(relevant options: heightf, innerf, outerf)
There are 3 configuration file options that control where a track will appear. Each track is defined as the area between two concentric circles, where the distance between those circles and their individual radii are specified by heightf, innerf, and outerf, respectively. These three options are not independent: given any two of them the third can be calculated according to the following very simple equation:
heightf = outerf - innerf
In practical terms what this means is that if you specify all three of them in the configuration file then Circleator will ignore one of them. If you choose not to supply any of these options then the Circleator will assign a default heightf value to the track, which determines how thick the track will be, and it will set the outerf value so that the track appears just inside the track that preceded it in the configuration file. So if the configuration file contains a number of tracks with no positioning or size information then by default the first track in the file will appear at the very edge of the circle (outerf = 1.0) and successive tracks will be nested inside those that came first. If too many tracks are placed using this method it is possible to run out of space in the circle (i.e., for a track to have a computed outerf value < 0), in which case Circleator will report an error when it tries to draw the figure.
The following figure shows a number of tracks that are labeled with their innerf - outerf values. It also illustrates how one track can be drawn on top of another by using transparency and an innerf - outerf range that overlaps with one or more other tracks:
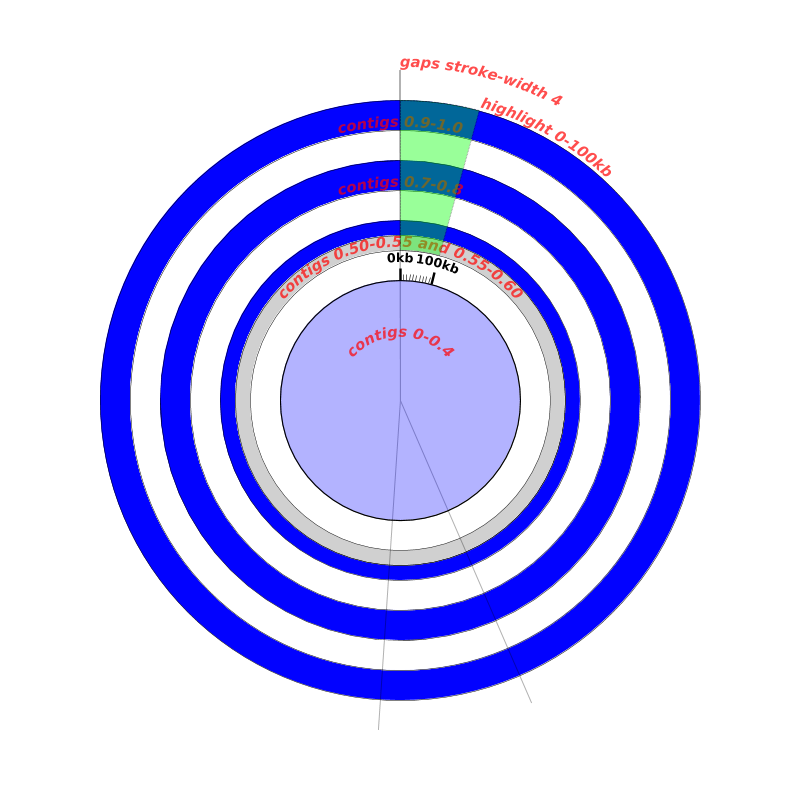
Here is the configuration file for the above figure: tracks-1.cfg
Going roughly from outermost to innermost the tracks in this figure are:
- the 3 thin grey lines indicating gaps are all in a track from 0-1.1 i.e., innerf=0, outerf=1.1, heightf=1.1: this track overlaps with all of the other tracks
- outermost blue “contigs” track from 0.9-1.0 i.e., innerf=0.9, outerf=1.0, heightf=0.1
- middle blue “contigs” track from 0.7-0.8 i.e., innerf=0.7, outerf=0.8, heightf=0.1
- thin blue “contigs” track from 0.55-0.6 (i.e., innerf=0.55, outerf=0.6, heightf=0.05
- thin grey track from 0.5-0.55 i.e., innerf=0.5, outerf=0.55, heightf=0.05
- green transparent track from 0.5-1.0 i.e., innerf=0.5, outerf=1.0, heightf=0.5: this track overlaps with all of the above tracks
- inner blue circle from 0-0.4 i.e., innerf=0, outerf=0.4, heightf=0.4
In this example one of the tracks (the gap track) has an outerf
value of 1.1. There’s nothing wrong with using values greater than 1,
but at some point these tracks will run off the edge of the
figure. How soon that happens is a function of the --pad
command-line argument, which determines how much blank space the
Circleator will leave on each side of the circle. By default the
circle has a diameter of 2400 and a --pad value of 400. Note also
that any of these three track configuration options (innerf,
outerf, and heightf) can be set to the special value same,
which means that they will take on the same value as the track that
immediately preceded the current track in the configuration
file. Setting both innerf and heightf to same, for
example, will create a track that exactly overlaps the one before
it. One can also add or subtract a constant value from same to
specify a value that is relative to the previous track. For example,
setting innerf=same+0.1 specifies an inner fraction that is 0.1
larger than the inner fraction of the previous track.
Feature Selection
(relevant options: feat_type,feat_strand, and others)
Many track and glyph types, for example the commonly-used rectangle
glyph, operate on sets of features. For example, the genes track is
defined thusly:
new genes rectangle 0.07 . . . gene . #000000
Note the value of gene in the feat_type field and “.” in the
feat_strand field; this indicates that a black (#000000) curved
rectangle (the rectangle glyph) should be drawn for every input
feature with type “gene”, regardless of strand. The definition for
the genes-fwd track is the same but with feat_strand set to “1”
to draw only forward-strand genes:
new genes-fwd rectangle 0.07 . . . gene 1 #000000
As usual, the feat_type and feat_strand may be included in the final options field rather than using the equivalent positional options. Note that either dashes or underscores may be used in the final options field: feat-type and feat_type are treated the same:
new genes-fwd-too rectangle 0.07 feat-type=gene,feat-strand=1,color1=#000000
Feature Filter Track Options
feat_type and feat_strand are both examples of feature “filters”. That is, they filter or restrict the set of features on which the track will operate. The following list enumerates the other feature filters that Circleator supports:
-
refseq-name - Filters out all features that are NOT from the specified sequence/contig
# draw only genes on contig239 new genes rectangle heightf=0.05,feat-type=gene,refseq-name=contig239 -
clip-fmin, clip-fmax - Filters out all features that are outside the specified sequence coordinate range
# label all genes in the region between 265kb and 275kb with their locus_ids: small-label label-type=spoke,innerf=1.04,feat-type=gene,label-function=locus,clip-fmin=265000,clip-fmax=275000 -
feat-type - Filters out all features that are NOT of the specified type (e.g., exon, CDS, gene, rRNA, tRNA)
# color all tRNAs red new genes rectangle heightf=0.05,color1=red,feat-type=tRNA -
feat-type-regex - Filters out all features whose type is NOT matched by the specified Perl regular expression
# color all tRNAs AND all rRNAs red new genes rectangle heightf=0.05,color1=red,feat-type-regex=[tr]RNA -
feat-strand - Filters out all features that are NOT on the specified strand
# color all forward-strand tRNAs red new genes rectangle heightf=0.05,color1=red,feat-type=tRNA,feat-strand=1 -
feat-min-length, feat-max-length - Filters out all features whose length (i.e., max genomic coord - min genomic coord) is less than (or more than) the specified value
# label all genes that are more than 20kb long: small-label label-type=spoke,innerf=1.04,feat-type=gene,label-function=locus,feat-min-length=20000 -
overlapping-feat-type - Filters out all feature that do NOT overlap with another feature of the specified type
# label all CDS features that contain at least one SNP new GL label feat-type=CDS,innerf=1.32,heightf=0.02,label-function=product,label-type=spoke,packer=none,overlapping-feat-type=SNP -
feat-tag - Used in conjunction with the remaining filters to select features based on their attributes. If none of the other feat-tag filters are given then it will filter all features that do NOT have the specified attribute.
-
feat-tag-value - Filter out all features whose feat-tag attribute does NOT have the specified value
# draw only SNPs with SNP_num_diffs=7 (i.e., SNPs at which 7 of the query genomes differ from the reference) new SNP rectangle 0.03 feat-type=SNP,stroke-width=1,color1=#000000,feat-tag=SNP_num_diffs,feat-tag-value=7 -
feat-tag-min-value, feat-tag-max-value - Filter out all features whose feat-tag attribute is less than (or more than) the specified value
# highlight genes whose expression value in the lung-derived sample is greater than 2 new H1 rectangle innerf=0.32,outerf=1.13,opacity=0.5,color1=#d0d0d0,color2=#000000,feat-type=CDS,feat-tag=EXP_Lung,feat-tag-min-value=2 -
feat-tag-regex - Filter all features whose feat-tag attribute does NOT match the specified Perl regular expression
# highlight genes whose gene product matches either "hypothetical protein" or "hypotheticalprotein" new HCH rectangle feat-type=CDS,feat-tag=product,feat-tag-regex=hypothetical\sprotein,innerf=same,outerf=same,opacity=0.8,color1=#00ff00
Feature Source
In all of the preceding examples we are implicitly considering all
of the genomic sequence features that appear in the original input
files (i.e., the file(s) passed to the --data and/or --contig_list
command line options) and selecting a subset of those features based
on feature type, feature strand, feature attribute values, etc. If the
original input file is a GenBank flat file, for example, then features
of the following types are common (this is not meant to be an
exhaustive list): source, gap, gene, CDS, tRNA,
rRNA, ncRNA, tmRNA, misc_feature, misc_binding. In
addition to features that appear explicitly in the input file(s),
Circleator also adds some features to represent the genomic
sequences/contigs and the spaces between them:
- reference_sequence - Circleator will always create a single feature of type reference_sequence that spans the entire reference coordinate system, including gaps if there are multiple sequences.
- contig - Circleator will create a feature of type ‘contig’ for each input sequence specified by the
--data,--sequence, or--contig_listoptions. - contig_gap - If the input contains multiple contigs then Circleator will create a feature of type ‘contig_gap’ between each pair of adjacent contigs (even if
--contig_gap_sizeis set to 0). If the--contig_listoption is used then the location and size of individual gaps can be specified explicitly in the contig list file. - genome - Circleator will add a feature of type ‘genome’ for each ‘genome’ line in the contig list file specified by
--contig_list. See the command line documentation for more information on the use of this option.
With this in mind, here are the possible feature sources for any given track:
-
The combination of all the features from all of the input files plus the special features mentioned above.
This is the default feature source. -
The set of features that are used by another track in the figure.
This feature source can be selected with the feat-track option, which specifies the (hopefully unique) name of the track whose feature list should be used. A common use of this option is to define a track that prints a label for every feature that was drawn in a previous track. Using the feat-track option is essentially a shortcut to avoid having to repeat all of the same feature filters in multiple tracks. Here is a simple example:# we use the predefined track type 'contigs' to show all features of type contig # (i.e., feat-type=contig) and name it "c1" contigs c1 # label only the contig features that were selected by track c1: medium-label feat-track=c1,label-function=primary_id,packer=none,innerf=same+0.01,outerf=same,text-color=#ffffff - The set of features loaded from a (new) external file.
The feat-file and feat-file-type options allow one to load features from files that were not included in the--data,--sequence, or--contig_listcommand-line options that were given when Circleator was run. The feat-file option specifies the full path to the file to be loaded and the feat-file-type option specifies what type the file is. If the file is a format accepted by BioPerl (e.g. GenBank flat file, multi-FASTA) then feat-file-type may be omitted. Otherwise it must be one of the following (case-insensitive) options:- ucsc_refGene
- ucsc_refGene_exons
- ucsc_knownGene
- ucsc_knownGene_exons
- ucsc_rmsk
- cufflinks_gtf
- skirret-snp
- merged-table-snp
- snp-table
- VCF
- csv-snp
- tabbed-snp
- trf
- gff
For example:
# Load SNPs for ATCC_30222 from VCF: new r1 rectangle 0.06 feat-file=data/ATCC_30222.extra-filtered.vcf,feat-file-type=VCF,feat-type=SNP,color1=snp_type,color2=snp_type,snp-query=ATCC_30222 - A single user-defined feature.
It is also possible to define a single simple feature directly in the configuration file by using the following track options:- user-feat-fmin, user-feat-fmax - coordinates of the feature in chado-style 0-indexed interbase coordinates
- user-feat-start, user-feat-end - coordinates of the feature in BioPerl-style 1-indexed base-based coordinates
- user-feat-strand - strand of the user-defined feature, either 0,1, or -1
- user-feat-type - type of the user-defined feature (e.g., “gene”, “new_feat_type_x”)
- user-feat-seq - reference sequence/contig on which the feature is localized
- user-feat-id - unique id for the user-defined feature
This can be useful for defining regions of interest directly in the configuration file. For example, the following configuration file excerpt highlights the user-defined sequence region from 0-5kb:
new SD5000 rectangle innerf=0,outerf=1.2,opacity=0.15,color1=#ff00ff,user-feat-fmin=0,user-feat-fmax=5000One could also define several user-defined features (at most one per line) with the same user-feat-type and then refer to all of those features in a subsequent track using feat-type. For example, here we create several ROI (region of interest) features and then highlight them. Note the use of the load glyph/track type, which does not draw anything, but only loads and/or creates new features:
new uf1a load user-feat-fmin=3898103,user-feat-fmax=3898620,user-feat-type=roi new uf2a load user-feat-fmin=844688,user-feat-fmax=845220,user-feat-type=roi new highlight_rois rectangle feat-type=roi,innerf=0,outerf=1.1,opacity=0.4,color1=red,color2=black
NOTE: Certain track types can be thought of as having both “input”
and “output” features. That is, they select a subset of the available
features by using feature filters (the input) and then they produce a
new set of features (the output) by processing the input features in
some way. For example, the compute-deserts track type takes a set
of input features and then creates a new “desert” feature wherever it
finds a large region that is empty of the input features. It could be
used to create a feature of type “SNP_desert” wherever there is 15kb
or more of sequence that does not contain any SNPs: the SNPs are the
input features and the SNP_deserts are the output features. Currently
the feat-track option always refers to the input features for a
track, never the output features. In the preceding example one could
instead refer to the output desert features by using the feature
filter feat-type=SNP_desert. Also in some cases the feat-type
option is used to specify the type of features that a track should
create, rather than filtering the input features.
Colors
(relevant options: color1, color2, text-color)
Wherever it expects a color specification (e.g., in the color1, color2, and text-color options) Circleator will accept any of the following:
-
Any valid SVG 1.1 color specifier
For example, all of the following are valid ways to set color1 to red:new cr1a rectangle feat-type=gene,color1=rgb(255,0,0) new cr1b rectangle feat-type=gene,color1=red new cr1c rectangle feat-type=gene,color1=#ff0000 -
Any color function defined in Circleator’s Circleator::FeatFunction::Color package:
- expression_level - Assigns color based on expression level and
uses the following additional options:
- sample - the sample whose expression level should be used to determine the color.
- exp-default-color - a default color to use if one is not specified by the following options.
-
exp-thresholds - a “ “-delimited list of one or more threshold values. -
exp-colors - a “ “-delimited list of colors, one for each value in exp-thresholds.
For example:
# Genes whose expression value is between 0 and 2 in the lung sample are colored green, those with value >2 are red: new genes rectangle 0.08 feat-type=CDS,color1=expression_level,color2=expression_level,sample=Lung,exp-default-color=black,exp-thresholds=0|2,exp-colors=green|red - regex_list - Assigns color based on matching a feature’s attribute to one or more regular expressions. Uses
the following additional options:
-
color[12]-regexes - a “ “-delimited list of Perl regular expressions against which to match the named feature attribute. -
color[12]-colors - a “ “-delimited list of color specifies, one for each of the Perl regular expressions. -
color[12]-min-lengths - a “ “-delimited list of minimum feature length values, one for each of the Perl regular expressions. -
color[12]-max-lengths - a “ “-delimited list of maximum feature length values, one for each of the Perl regular expressions. - color[12]-attribute - the feature attribute to match against the regexes: currently “display_name” and “product” are the only options
- color[12]-default - the color to use for any feature whose display_name or product matches none of the regular expressions
For example:
# Attempt to color-code hypothetical genes based on their length (0-499,500-999,1000-1999,2000+): new genes rectangle 0.08 feat-type=CDS,color1=regex_list,color1-regexes=hypothetical|hypothetical|hypothetical|hypothetical,color1-colors=#375817|#ef3c17|#ef16b7|#162bef,color1-attribute=product,color1-default=none,color1-min-lengths=0|500|1000|2000 -
- snp_test - Assigns color based on whether a SNP/small sequence variant affects a single base or multiple bases. Uses
the following additional options:
- snp-query - the name of the query (i.e., non-reference) genome or strain in question
For example:
# Display the variant allele/genomic sequence bases from sequence AE015925.1, using text-color=snp_text to draw single base alleles white and multibase alleles gray new SNPL1 label 0.02 innerf=same,label-track-num=-1,label-function=snp_base,text-color=snp_text,snp-query=gi|29835126|gb|AE015925.1|,packer=none - snp_type - Assigns color based on the type of a SNP/indel. Uses
the following additional options (all of which except snp-query are optional and have predefined defaults):
- snp-query - the name of the query (i.e., non-reference) genome or strain in question
- snp-no-hit-color - color to use for SNPs/indels where the variant position could not be identified in the snp-query strain
- snp-same-as-ref-color - color to use for SNPs where the query sequence is the same as the reference sequence
- snp-unknown-color - color to use for SNPs that haven’t been classified as synonymous/nonsynonymous/intergenic
- snp-intergenic-color - color to use for intergenic SNPs (wrt to some reference annotation)
- snp-syn-color - color to use for synonymous SNPs
- snp-nsyn-color - color to use for nonsynonymous SNPs
- snp-multiple-color - color to use for SNPs that may be both synonymous and nonsynonymous, depending on context
- snp-ins-color - color to use for insertions in the query relative to the reference
- snp-del-color - color to use for deletions in the query relative to the reference
- snp-intronic-color - color to use for intronic SNPs
- snp-readthrough-color - color to use for readthrough SNPs
- snp-other-color - color to use for any SNP not covered by any of the other cases
For example:
# set snp-nsyn-color to 'none', which in this case has the effect of showing only the SYNonymous SNPs new snp2 rectangle heightf=0.07,feat-track=snp1,color1=snp_type_no_indel,color2=snp_type_no_indel,snp-query=PCTRA_SC110_consensus_hhcedit,snp-nsyn-color=none,snp-intergenic-color=none - snp_type_no_indel - Assigns color based on the type of a SNP/indel. Accepts the same options as snp_type, minus snp-ins-color and snp-del-color
Labels
(relevant options: label-text, label-function)
The label track type in Circleator allows labels (i.e., short text strings) to be drawn next to tracks and/or specific features. For example, one might label an entire track “%GC-Content” or one might label a highlighted region of interest “highly variable region 1” or one might label all tRNAs with their anticodon. There are two ways to specify what label should be drawn:
-
The label-text option specifies a literal string that the label should display. It is important to note that the current configuration file format does NOT allow for spaces in option values, although it is possible to use an underscore character or the string “\ ” (i.e., the HTML character entity reference for a non-breaking space) instead. So the following is NOT legal:
large-label label-text=C. albicansBut both of these are legal:
large-label label-text=C._albicans large-label label-text=C.&nbsp;albicans -
The label-function option can be used to specify any of the label functions defined in Circleator’s
Circleator::FeatFunction::Labelpackage. These label functions assign labels to individual features using a variety of methods. Some are specific to particular feature types whereas others should work regardless of type:- tag - this function can be used to extract any BioPerl-defined tag/attribute value from a feature. For example, it could be applied to extract the ID field from features read from a GFF3 file. The function supports the following options:
- tag-name - the name of the tag/attribute whose value is to be used for the label
- tag-value-separator - a string that should be used to join together multiple tag values in the case where a feature has more than one.
- tag-ignore-multiple-values - set this to 1 to ignore multiple values and use only the first.
For example:
new pl label innerf=same+0.02,outerf=same-0.025,label-function=tag,tag-name=ID,feat-type=p_arm,text-color=white,packer=none,label-type=spoke new ql label innerf=same,outerf=same,label-function=tag,tag-name=ID,feat-type=q_arm,text-color=white,packer=none,label-type=spoke - accession - labels features with their accession number, if any.
- display_name - labels features with their BioPerl
display_name(), if any. - id - labels features with their BioPerl
id(), if any. -
primary_id - labels features with ther BioPerl
primary_id, if any. -
locus - labels features with their BioPerl locus id, if any. Note that the following two lines are equivalent:
small-label label-type=spoke,innerf=1.12,feat-type=gene,label-function=locus small-label label-type=spoke,innerf=1.12,feat-type=gene,label-function=tag,tag-name=locus,tag-ignore-multiple-values=1 -
product - labels features with their BioPerl product, if any. Note that the following two lines are equivalent:
small-label label-type=spoke,innerf=1.12,feat-type=CDS,label-function=product small-label label-type=spoke,innerf=1.12,feat-type=CDS,label-function=tag,tag-name=product,tag-ignore-multiple-values=1 - bsr_count - labels a BSR feature with the number of genomes in which the corresponding gene is conserved according to the BLAST Score Ratio threshold. Supports the following options:
-
genomes - a “ “-delimited list of BSR query genomes to consider when counting the number of genomes in which each gene is conserved. - threshold - a BSR threshold value above which genes are considered to be conserved. Default is 0.4
For example:
new glc1 label 0.02 outerf=same,feat-track=bsr_track_e,label-function=bsr_count,text-anchor=middle,packer=none,label-type=curved,genomes=gcp8455|gcpCP3|gcpGR9|gcpM56|gcpMN|gcpNJ1|gcpVS225|gcpWC|gcpWSRTE30 -
- genomic_seq - labels a feature with its literal genomic sequence. Typically used for individual base features that have been expanded to the point where there’s room to display the corresponding A,C,G, or T within.
- length_bp - labels features with their length in base pairs.
- length_kb - labels features with their length in kilobases.
-
position - labels features with their genomic sequence position.
-
rRNA_product - labels rRNA features with their type (e.g., 16S, 23S).
- snp_base - labels SNP/indel features with their DNA sequence in a specified strain/genome. Supports the following options:
- snp-query - the name of the query (i.e., non-reference) genome or strain in question
- snp-no-hit-label - a specific label/string to use when the SNP position could not be identified in the specified query strain.
-
snp-same-as-ref-label - a specific label/string to use when the DNA sequence in the specified query strain is the same as the sequence in the reference strain. Setting this to \ , for example, will print the DNA sequence only for those loci that differ from the reference.
small-label label-function=snp_base,label-track-num=-2,text-color=#a0a0a0,style=default,snp-query=CP1041
- snp_ref_base - similar to snp_base, but it displays the reference DNA sequence at a given SNP position.
-
snp_gene_id - displays the gene_id (if any) associated with a SNP feature.
Note that the following two lines are equivalent:new SNPP label innerf=1.20,heightf=0.35,label-track-num=-2,label-function=snp_gene_id,label-type=spoke,text-anchor=start new SNPP label innerf=1.20,heightf=0.35,label-track-num=-2,label-function=tag,tag-name=gene_id,tag-ignore-multiple-values=1,label-type=spoke,text-anchor=start -
snp_product - displays the gene product (if any) associated with a SNP feature.
Note that the following two lines are equivalent:new SNPP label innerf=1.20,heightf=0.35,label-track-num=-2,label-function=snp_product,label-type=spoke,text-anchor=start new SNPP label innerf=1.20,heightf=0.35,label-track-num=-2,label-function=tag,tag-name=SNP_product,tag-ignore-multiple-values=1,label-type=spoke,text-anchor=start
- tag - this function can be used to extract any BioPerl-defined tag/attribute value from a feature. For example, it could be applied to extract the ID field from features read from a GFF3 file. The function supports the following options:
Opacity
(relevant options: opacity)
The opacity/transparency of any track can be set with the opacity option, which takes as its value a number between 0 and 1, with 0 meaning 0% opacity (i.e., the track is completely invisible) and 1 meaning 100% opacity (i.e., it completely obscures anything directly behind it). Values between 0 and 1 can be used to make tracks semi-transparent. For example, one might highlight a region of interest by overlaying a shaded area with opacity set to 0.3 or 0.4, like so:
new highlight1 rectangle user-feat-fmin=50000,user-feat-fmax=100000,color1=red,color2=none,opacity=0.3,innerf=0,outerf=1.1
Loops
(relevant track types: loop-start, loop-end)
Circleator supports the use of loops to simplify the construction of configuration files when the same track or set of tracks must be repeated many times for a different subset of the data (e.g., as when showing SNPs, gene clusters, or BSR data for a number of genomes or strains in a large multi-strain comparison.) Loops may be added to a configuration file using two special track types, loop-start and loop-end. At present a loop may specify only a single loop variable (i.e., a special keyword that will be replaced by each of a list of values), like so:
# highlight a set of genes:
new ls1 loop-start loop-var=GENE,loop-values=VC_0788|VC_A0970|VC_0915|VC_A1016|VC_A0790
# the contents of the loop, starting here, are repeated for each of the loop-values above:
# with each loop iteration the keyword <GENE> is replaced by each of the values in turn
new gi rectangle feat-type=gene,feat-tag=locus_tag,feat-tag-value=<GENE>,innerf=0,outerf=1.1,color1=red,color2=grey,opacity=0.2,stroke-width=1.5
medium-label innerf=1.12,feat-track=-1,label-function=locus,label-type=spoke
new le1 loop-end
Z-index
(relevant options: zindex )
The Z-index of a track determines its relative stacking position (i.e., top to bottom) in the figure. Currently the Z-index of each track is determined by its position in the configuration file, with the first track in the file appearing on the bottom of the stack and the last track in the file appearing on the top of the stack.
